- Latest News
- ‘Unique’ Omani blood disorders under global research microscope
11-11-2018
Times of Oman
Muscat: Many Omanis carry a rare blood disorder that could reduce the production of haemoglobin in the body and cause paleness, weakness, fatigue, and more serious complications, although not all carriers develop symptoms, a researcher in the Sultanate has revealed.
Dr Halima Al Balushi, a researcher and senior specialist at Oman’s Royal Hospital, conducted a study which reveals that a large percentage of Omanis have alpha thalassaemia and that five per cent have sickle cell anaemia.
Dr Al Balushi, discussing her research with the Ministry of Higher Education, added that Omanis also have a rare form of sickle cell disease which is being investigated by medical researchers all over the world. The condition is so rare it has been labeled “Oman type sickle cell disorder” and added that around 70 people in the Sultanate currently suffer from the condition.
Al Balushi, in an interview with the Ministry of Health, added: “Genetic blood disorders are prevalent in the Sultanate, which is why studies such as these can identify blood cells, as well as treatment methods. The study used inexpensive medications that proved effective, and also tested genetic treatments to satisfying results.”
“Thankfully, the Sultanate has an abundance of research labs that can aid in producing medications, and the University of Cambridge has shown interest in studying genetic blood disorders. This is also due to the grants of His Majesty, who has shown interest in scientific research,” she added.
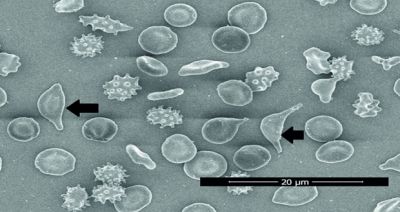
While alpha thalassaemia is a blood disorder that reduces the production of haemoglobin, thus impacting oxygen flow, Al Balushi studied samples of blood that contained the disorder known as Haemoglobin (Hb) S-Oman, also called Omani Sickle Cell Anaemia, which distorts the shape of red blood cells into a shape similar to that of a hat, and can cause extreme cases of pain.
Al Balushi added, “The study at the British Journal of Haematology revealed that the red cells in this anomaly are baffling, as they have all the characteristics of the disease and can cause medium to extreme levels of pain. These cells, nicknamed ‘Oman Type Sickle Cells’ are also rather rare, and have been recorded in only 70 persons in the Sultanate.”
According to the publication, HBS Oman is considered the second rarest form of sickle cell. Carriers can also develop extremely painful symptoms. Al Balushi explained that the genetic anomaly can also spread fast, as it can be inherited and does not require both parents to have the disorder.
The World Health Organisation has also raised awareness over the dangers of thalassaemia.
“Alpha thalassaemia causes hydrops fetalis and perinatal death, often with life-threatening obstetric complications for the mother, and prenatal diagnosis usually leads to termination of pregnancy,” said Bernadette Modell, a professor who specialises in thalassaemia, and Matthew Darlison, who is a member of the World Health Organisation’s Collaborating Centres (WHOCC). “Some cases have recently been saved by intrauterine transfusion, despite a high risk of severe mental and physical handicap.”
“Inherited haemoglobin disorders (sickle-cell disorders and thalassaemia) were originally characteristics of the tropics and subtropics but are now common worldwide due to migration,” they added. “Since they can be controlled cost-effectively by programmes that integrate treatment with carrier detection and genetic counselling, WHO has recommended the global development of these services. However, service development can be unexpectedly challenging, because it requires the inclusion of genetic approaches in health systems.
Modell and Darlison also shared methods of prevention of thalassaemia in the Sultanate. “A policy of detecting carriers and informing them of their risk, and possibilities for reducing it, usually leads to a fall in births and deaths of affected children,” they said. “Requirements are the same for thalassaemias and sickle-cell disorders. In most countries, the approach develops in three stages.”
“First, retrospectively informing parents with affected children of their 25 per cent recurrence risk allows them to limit the family size and, where average family sizes are typically large, this approach can significantly reduce affected birth prevalence,” they said.
“Second, introduction of prenatal diagnosis for couples with affected children enables them to have a family, but has little further effect on affected birth prevalence. Access may also be limited by economic, medical, social or legal factors. Third, information and prospective carrier screening is provided for the whole population.”





